MICHAEL SCHAEFER
Quality Management and Regulatory Affairs in Medical Devices

Trainings
writer?")
Freelancing and technical (ghost)writing

Work-life balance
Trainings
Maintain your qualifications
Freelancing
Get supported
All Downloads
Download free materials
Work-life balance
Charging up energy

So what is the difference?
Quality-on-site.com is a platform for quality practitioners provided by Michael Schaefer, Expert for Quality Management and Regulatory Affairs in Medical Devices. It is my goal to provide and exchange valuable information and knowledge needed for continuously improving quality methods in medical device industries to reduce patient risk, increase quality and fulfil regulatory requirements. Training and support is available upon request.
For more information and networking contact info@quality-on-site.com
The information on this site provided is free; however the author does not take any responsibility for contents and correctness.
My skill set
A quality system on its own will not improve product or process quality. Applying suitable tools and methods are crucial to ensuring long-term success and customer satisfaction. Over the past years I was privileged to develop further my own skillset to implement both compliant and cost-efficient quality systems by focusing on patient safety and error prevention. Take a look into my skill set!

The latest (LinkedIn) Gossip
Celebrating 2025/2026
At the end of the year, I am grateful for the journey: celebrating 30 years in industry, 25 years in medical devices, 15 years as a trainer, and 10 years as an authorized auditor. Thankful for the people, learning, and opportunities along the way.
New trainings available!
My updated training plan is available and I am proud to present some new trainings! Looking forward seeing you in the remaining 2025 and/or new 2026!
New MDSAP website
MUST-READ! The new MDSAP website is available including all relevant documents. Finally you can now access as well the new revision 002 of MDSAP AU P0037.002 NC grading.
Benefit-Risk under MDR 2017/745
Nach einer intensiven Vorbereitung freue ich mich sehr mit der TÜV SÜD Akademie ein neues Seminar starten zu können. Unser Seminar hebt die Nutzen-Risiko-Analyse als Herzstück der Konformitätsbewertung unter der MDR 2017/745 hervor. Obwohl diese Analyse nun als unverzichtbar gilt, fehlen hä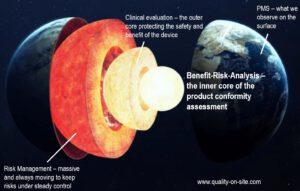 ufig konkrete Vorgaben für Umsetzung und Dokumentation. In unserem eintägigen Lehrgang lernen Sie nicht nur die Grundlagen der Nutzen-Risiko-Analyse kennen, sondern auch die verschiedenen Aufgaben und Zusammenhänge des Risikomanagements gemäß ISO 14971 in Verbindung mit der klinischen Bewertung. Wir sind sehr gespannt, ob wir Ihnen eines der schwierigsten Themen der MDR praxisnah vermitteln und somit etwas Licht ins Dunkel bringen können.
ufig konkrete Vorgaben für Umsetzung und Dokumentation. In unserem eintägigen Lehrgang lernen Sie nicht nur die Grundlagen der Nutzen-Risiko-Analyse kennen, sondern auch die verschiedenen Aufgaben und Zusammenhänge des Risikomanagements gemäß ISO 14971 in Verbindung mit der klinischen Bewertung. Wir sind sehr gespannt, ob wir Ihnen eines der schwierigsten Themen der MDR praxisnah vermitteln und somit etwas Licht ins Dunkel bringen können.
Audit hint: impact assessment of changes
Audit hint: Please make sure that you document a proper impact assessment of changes. Especially for design changes you must evaluate, for example, impact to shelf-life, bicompatibility, and sterilization. If you are not able to provide such assessments during the audit it might remain unclear, if the modified product will remain as safe as the previous version. The impact assessment must also conclude if regulatory activities are required.
Revalidation of software
Audit hint: Validation of software used within the quality management system as per ISO 13485 is not new and most of the companies did their homework. However, keep in mind that any change to such software must be assessed if revalidation is required. Just recently I audited a software which was validated two years ago and a major release was conducted in the meanwhile without any documentation.
Medical device recalls in Canada
Changes to Canadian medical device regulation with respect to recalls: get your procedures ready for Dec 17, 2024. Make sure that the revised requirements are implemented. HC offers a nice flowchart guiding along the recall procedure. For voluntary recalls, your communication to HC will start 24h after making the decision to recall, on the day on the recall and 30 days after completion. Type III recalls will not need to be reported.
Address
Heiligkreuzstrasse 59,
72379 Hechingen Germany